
Introduction
신약개발에서 진행되는 연구 중 MHC(Major Histocompatibility Complex, 주조직 적합성 복합체)와 펩티드(peptide)의 결합 친화도(binding affinity)를 예측하는 연구 분야가 있다. 먼저 MHC 같은 경우는 면역 반응에 관여하는 당단백질로, 대표적으로 백혈구(HLA, Human Leukocyte Antigen)가 있다. 펩티드의 경우 단백질과 동일하게 아미노산으로 구성되지만 단백질을 결정짓는 3차원 얼개가 없는 것이 가장 큰 차이점이다. 그렇다면 MHC-peptide의 binding affinity 예측은 왜 중요할까? 이는 MHC와 펩티드 간 상호작용하는 그 기전에 답이 있다. 결론은 향후 신약을 만들었을 때의 우리 몸의 면역 반응을 판단하기 위함이다.
MHC-peptide 면역학적 상호작용
MHC는 항원으로부터 떨어져 나온 펩티드 조각을 인식하는 능력을 갖고 있다. MHC는 세포표면에 자신이 만든 그루브에 펩티드 조각을 꽂아두면 면역 반응에 관여하는 T 세포가 와서 T 세포 수용체로 면역원(펩티드)을 인식하고, 만약 악성이라 판단될 경우 항체를 만들라는 면역 반응을 일으키게 된다. 하지만 이와 같은 기전이 일어나기 위해서는 MHC와 펩타이드 둘 간의 충분한 binding affinity로 결합이 되어야만 T 세포가 활성화 될 수 있다. 그렇기에 binding affinity를 예측하는 것은 이러한 면역학적인 과정을 이해하는 데 한 걸음 나아가는 것과 같고, 향후 신약 개발에 있어 후보 물질이 될 수 있는 펩티드를 스크리닝하는 데 많은 도움이 된다.
펩티드의 장점과 단점
참고로 펩티드 의약품은 분자 구조상 독성 문제가 적고, 조직 내 축적량이 적어 신약 후보물질로 주목 받는다. 하지만 체내에서 쉽게 분해된다는 단점이 있어 체내 안정성 확보가 어렵고, 이러한 펩티드 후보 물질을 탐색하는 시간이 오래걸린다는 문제가 존재한다.
MHC-peptide binding affinity 예측 연구의 과거와 현재
연구의 흐름은 in-vitro 방식에서 in-silico 방식으로 바뀌었다. 과거에는 일일이 MHC-peptide의 binding affinity를 계산했다. 하지만 이는 수많은 시간과 자원이 소모되었는데 그 이유는 MHC의 특성상 매우 큰 다형성(polymorphism)을 가졌기 때문이다. 다형성으로 인해 MHC와 펩타이드 시퀀스 간의 여러 경우의 수를 다 고려할 수 없었고, 따라서 computational 접근으로 나아가게 되었다. 이 computational 접근이 in-silico 방식의 또 다른 이름이다. in-silico 방식으로 나아갈 수 있게 된 배경은 in-vitro 방식을 통해 MHC-peptide의 binding affinity에 대한 데이터가 쌓였고, 아래와 같은 데이터셋이 축적되며 가능해졌다.
위와 같은 데이터셋들은 MHC-peptide binding affinity에 관한 아래의 주요 데이터베이스들에 모이게 되었다.
참고로 현재에도 대표적으로 가장 많이 사용되는 데이터베이스는 IEDB이다. in-silico 방식에는 위의 데이터베이스를 활용하는 크게 두 가지 방법이 있다. 첫 번째는 allele-specific 방법이고, 두 번째는 pan-specific 방법이다. 참고로 allele란 대립유전자를 의미하는 것으로 지금의 문맥에서는 인간의 MHC인 HLA를 의미한다. 아래는 두 방식에 대한 그림이다.
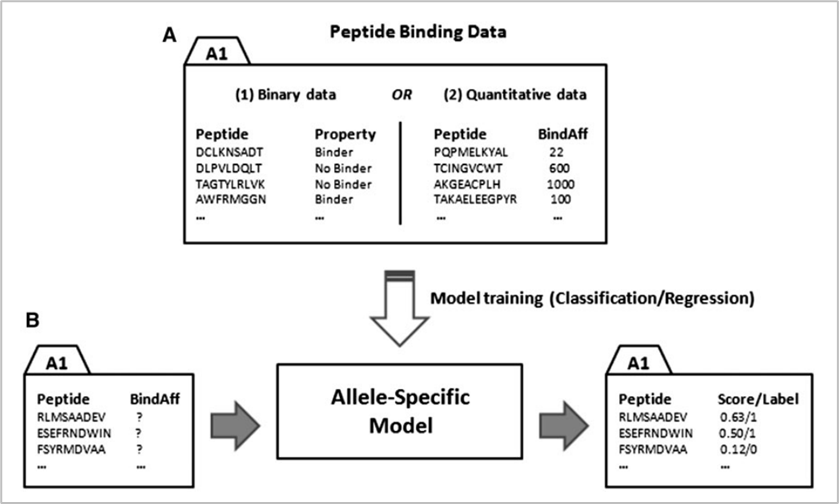
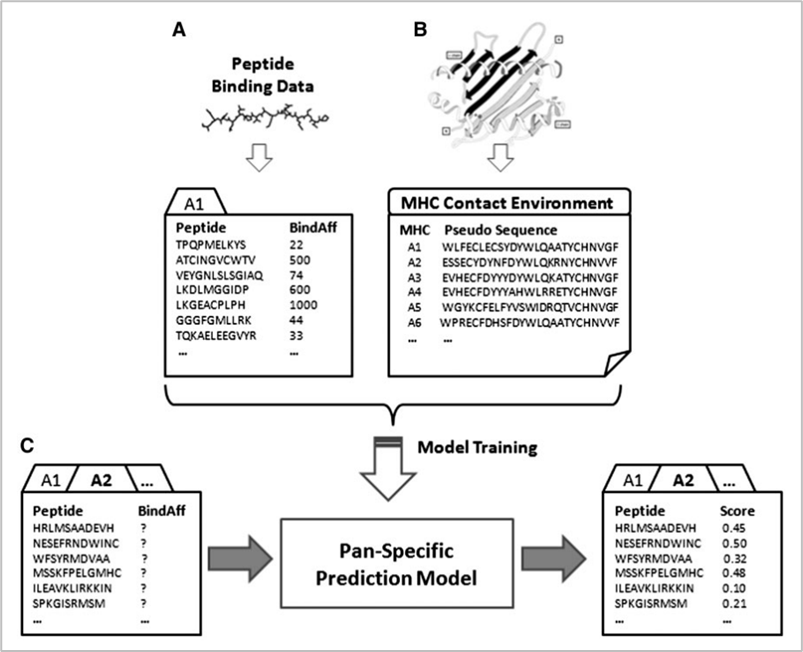
두 방식의 공통점은 머신러닝 모델을 만들어 예측하는 것이다. 핵심 차이점은 allele-specific은 하나의 allele당 하나의 모델을 만들어야 하는 반면 pan-specific 방법은 하나의 모델에 여러 allele를 학습시킬 수 있다. 때문에 allele-specific 방법은 범용성은 약하지만 특정 allele와 peptide가 어떤 binding affinity로 결합할지에 대해 pan-specific에 비해 비교적 더 정확하게 예측할 수 있다는 장점이 있다. 반면 pan-specific 모델은 특정한 allele와 peptide의 binding affinity 예측에는 약하지만, 전반적으로 일반화된 성능을 낼 수 있다는 것이 장점이다.
하지만 최근에는 증가하는 데이터셋과 머신러닝 모델 구조로 인해 allele-specific 모델보다 pan-specific 방식이 더 많은 각광을 받는 추세이며, 나아가 pan-specific 모델에 transfer learning을 적용함으로써 allele-specific 모델의 장점을 함께 취하는 방식도 사용되고 있다.
MHC-peptide binding affinity 예측 도구
MHC-peptide binding affinity를 예측 하는 도구들은 1990년대 말 이후 지금까지도 관련 연구자들에 의해 만들어지고 있다. 아래는 2011년 기준으로 사용되고 있는 pan-specific 모델을 정리한 표이다.
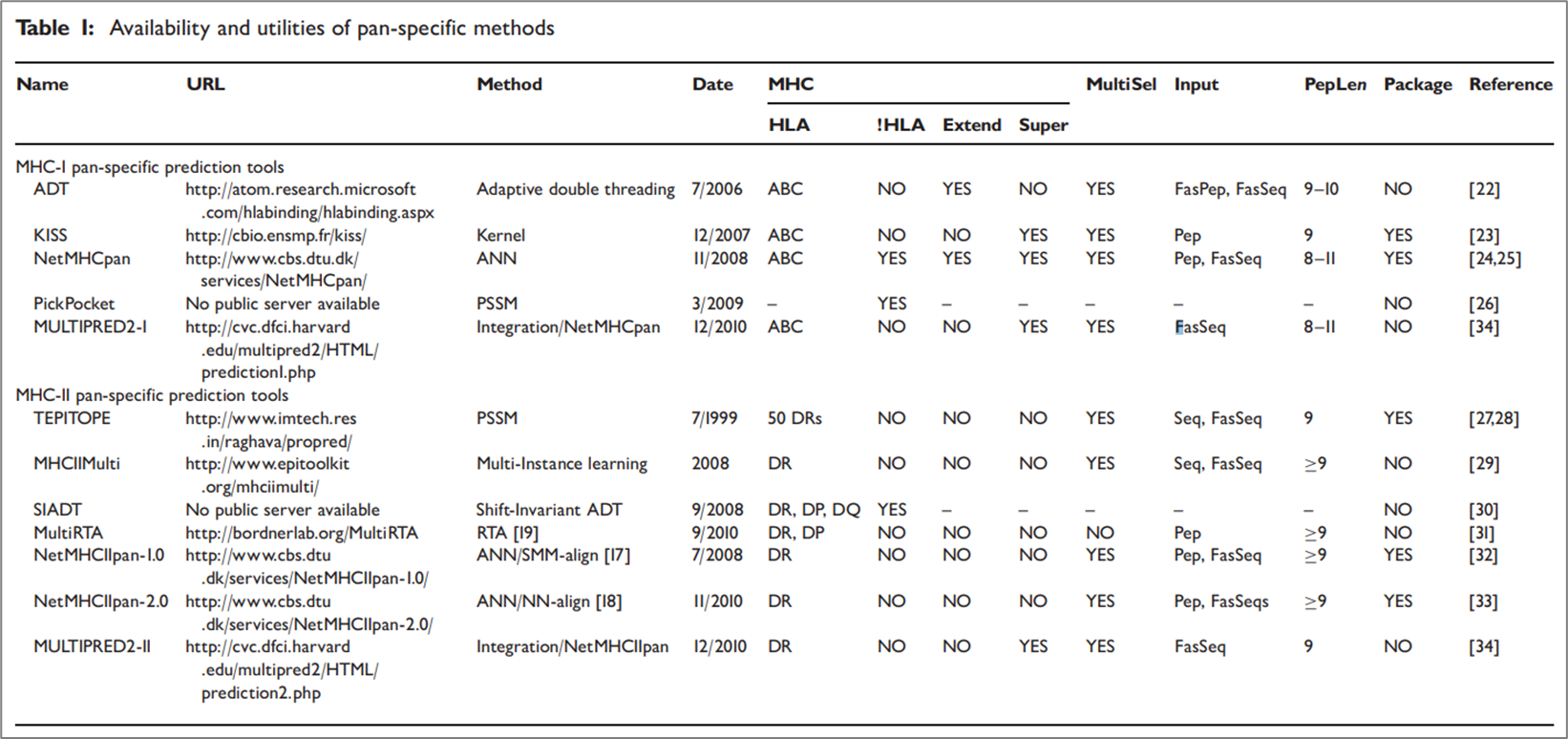
참고로 MHC는 크게 3가지 클래스가 존재한다 MHC-I, MHC-II, MHC-III이다. 하지만 연구는 주로 MHC-I과 MHC-II를 기준으로 이루어진다. 위 그림을 크게 MHC-I pan-specific 예측 모델과 MHC-II pan-specific 예측 모델로 나눌 수 있는데, MHC-I에는 버전이 업그레이드되어 지금도 좋은 성능을 보이고 있는 NetMHCPan이 대표적이라 할 수 있다. 현재에는 더 발전하여 이외에도 ACME, DeepSeqPan, DeepAttentionPan, 등의 모델이 있다. MHC-II 같은 경우도 마찬가지로 NetMHCIIpan 모델이 대표적이라 할 수 있고 현재에는 더 발전하여 이외의 모델들이 여럿 존재한다.
MHC-펩티드 피처화 방안 & 머신러닝 모델 종류
allele-specific 모델과 pan-specific 모델은 학습을 위해 MHC와 펩티드를 피처화 시키는 과정이 중요하다. 머신러닝을 사용하지 않고 계산했던 과거에는 Scoring Matrix를 사용했고 동작은 다음과 같다.

축적된 MHC-peptide에 대한 데이터를 기반으로 BLOSUM62와 PSSM이라는 대표적인 scoring matrix를 사용했다. 이는 펩티드의 motifs를 찾으려는 시도로, motifs는 특징이 보존된 짧은 펩티드 서열을 의미한다. scoring matrix를 만들기 위해 펩티드의 물리 화학적인 특성이나 한 아미노산에서 다음 아미노산으로 어느 정도의 빈도로 바뀌는가 또는 시퀀스의 유사도를 계산한 값을 반영한 matrix라 할 수 있다. 이를 통해 최종적으로 MHC(HLA)와 펩티드 시퀀스가 어느 정도의 binding affinity로 결합할 것인지 예측하는 것이다. 2019년 기준 대표적인 도구 리스트는 다음과 같다.
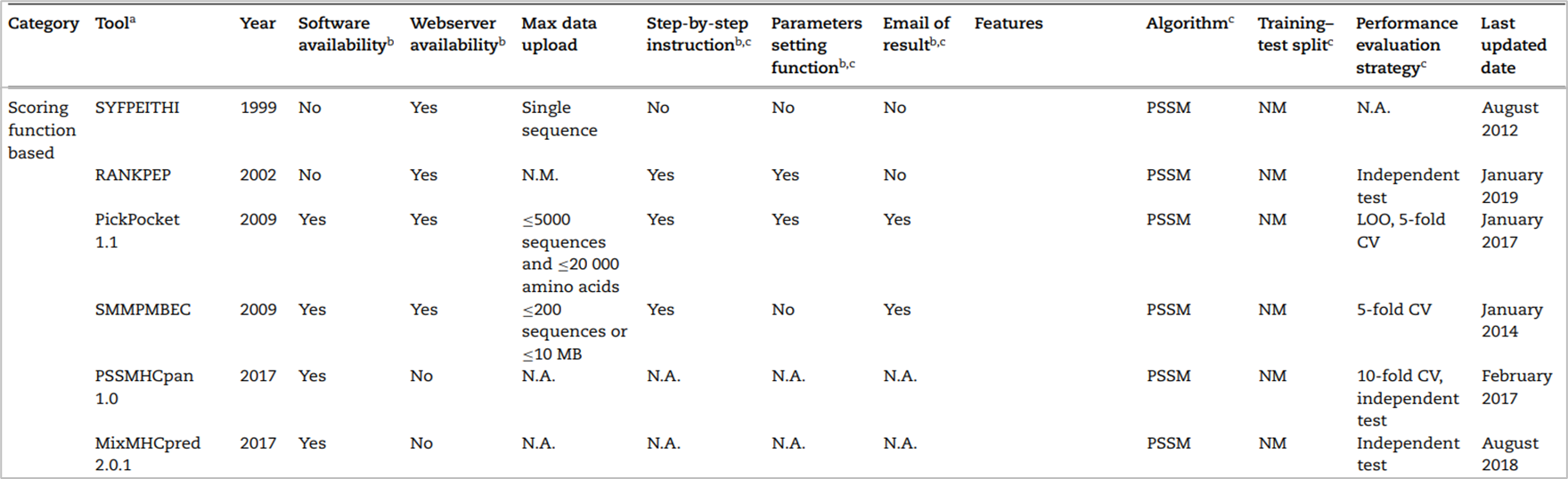
하지만 이 방법에도 단점이 있었으니, 정적인 scoring matrix를 통해서는 문제를 선형적으로 풀 수 있을 뿐 동적이고 비선형적으로 풀기 어렵다는 것이다. 이러한 한계점으로 인해 머신러닝 기반의 방법이 사용되었다.
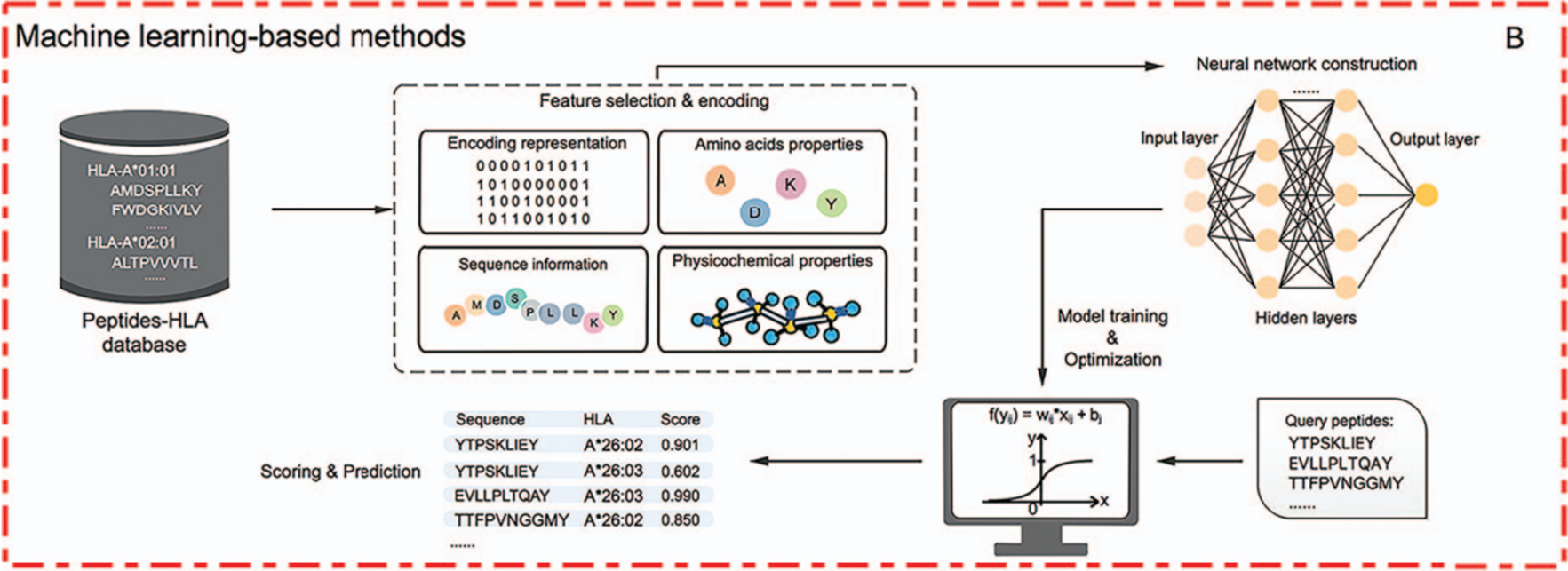
기존 scoring function 기반 방법과의 차이점은 뉴럴넷에 학습을 시킨다는 것이다. 하지만 동일한 것은 여전히 피처화 시키는데 있어서 BLOSUM62와 PSSM, one-hot encoding과 같은 방법을 사용한다. 데이터셋으로는 in-vitro 방식 또는 이전 연구들에서 binding affinity가 실험적으로 검증된 것을 사용하며 결과적으로 펩티드를 binder 또는 non-binder로 분류하는 것이 특징이다. 2019년 기준 대표적인 도구리스트는 다음과 같다.
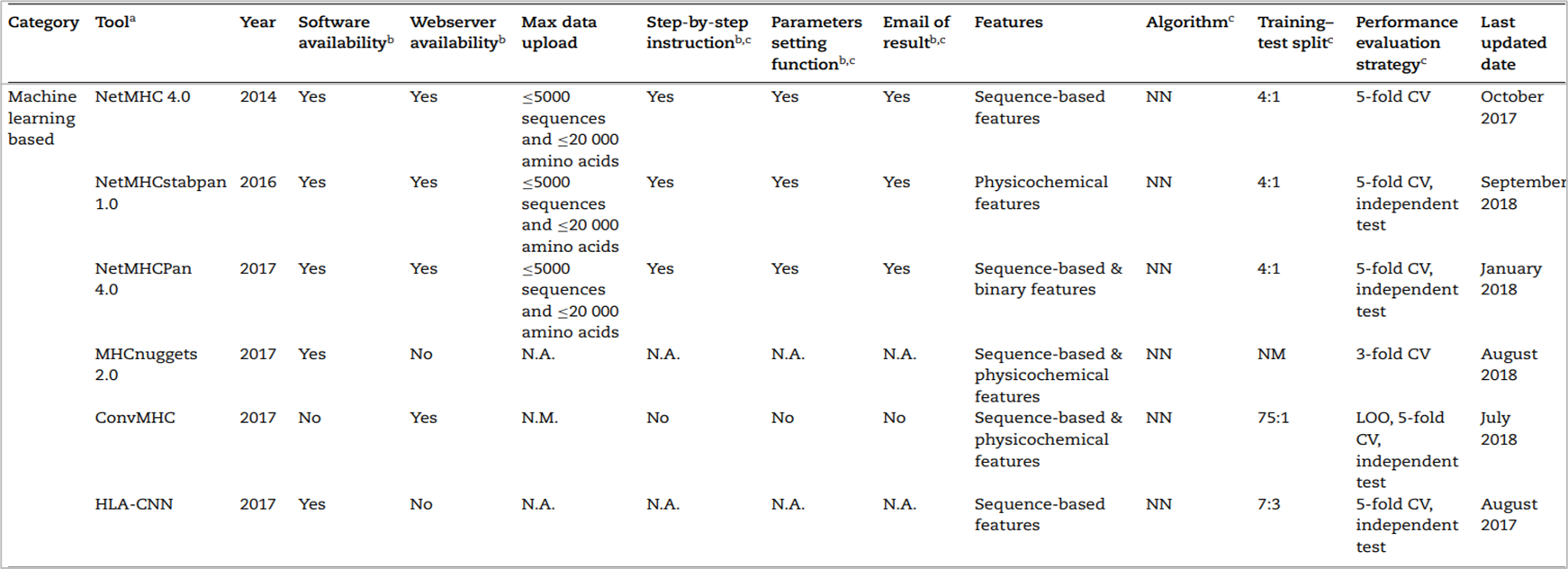
그 중 가장 대표적인 모델은 NetMHCPan4.0이 있고, 후속 연구들에서 비교 대상으로 자주 사용된다.
마지막으로는 consensus 방식으로 scoring function 기반 방법과 machine learning 기반 방법을 결합한 것이다.
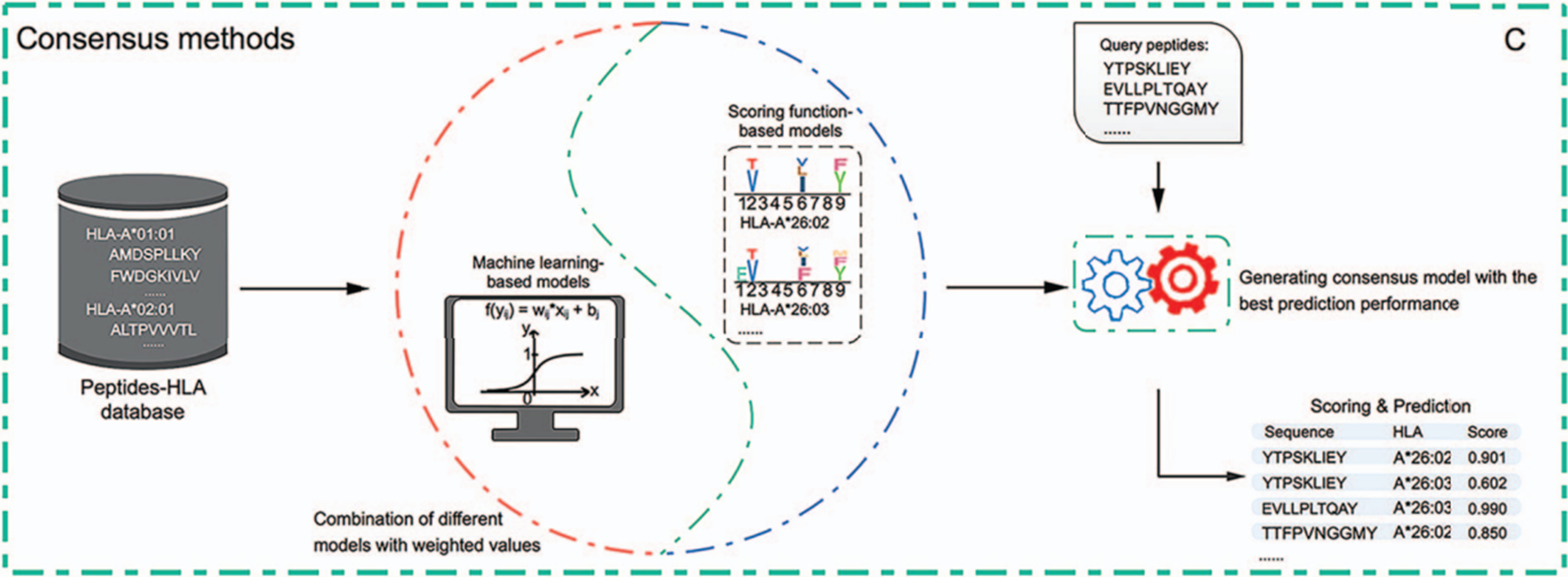
정확히는 scoring function 기반의 모델과 machine learning 기반의 모델의 출력값에 가중치를 부여하여 앙상블하는 방법이다. 예를 들면 가장 믿을 만한 모델의 예측 값에는 N점, 조금 낮은 모델의 예측 값에는 N-1점, ...을 부여하여 가중합을 매기고 평균을 구하는 것이다. 패러다임 시프트가 없는 이상 위와 같은 방법이 계속 사용될 것으로 전망된다. 2019년 기준 대표적인 도구 리스트는 다음과 같다.
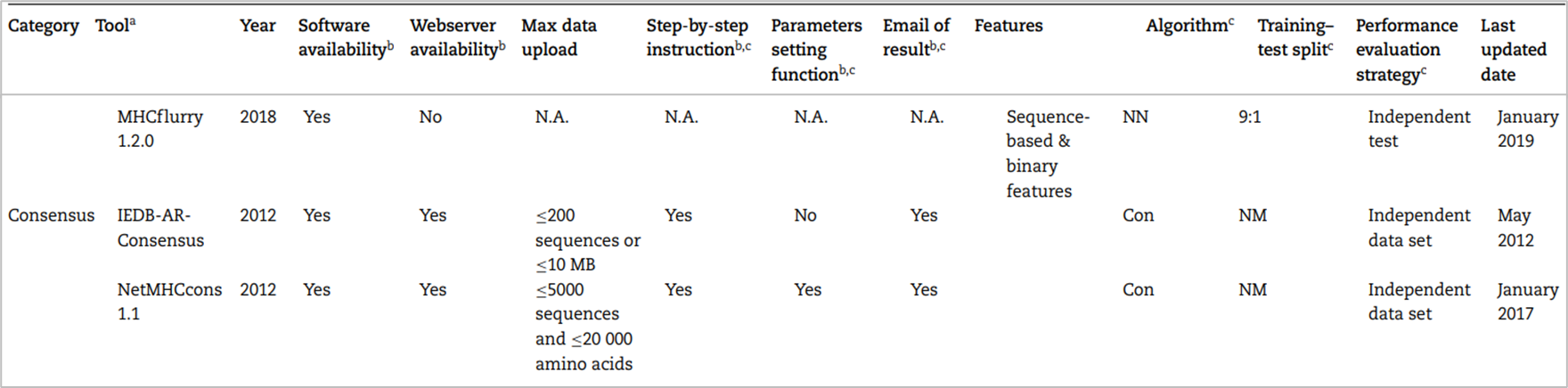
가장 최근에 나온 MHCflurry1.2.0이 가장 대표적인 도구일 것이다.
여기까지 신약개발의 연구 분야 중 하나인 MHC-peptide binding affinity prediction의 연구 동향을 살펴보았다. 이 연구 분야에 대해 알아볼 때 생각보다 국내 자료가 많이 없어 빠른 파악이 힘들기도 했기에 관련 연구를 지향하는 사람들에게 작은 도움이 되었으면 한다.
Reference
[1] Toward more accurate pan-specific MHC-peptide binding prediction: a review of current methods and tools
[2] A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction
'Science > 생물학' 카테고리의 다른 글
| [세포생물학] 센트럴도그마: 전사와 번역 (Transcription and Translation) (0) | 2022.05.13 |
|---|---|
| [생화학] 아미노산과 펩티드 결합 (0) | 2022.04.28 |
| [생명공학] DNA의 정보처리과정에서 오류율이 적은 이유 (0) | 2021.10.21 |